Pompeho nemoc, přesně označována jako glykogenóza typu II, je velmi vzácné autozomálně recesivně dědičné onemocnění. Příčinou onemocnění je mutace genu pro lyzozomální kyselou α-1,4 glukosidázu (GAA) – enzym, který je odpovědný za štěpení α-1,4 vazby v glykogenu. Pokud je tento enzym v lidském organizmu deficitní, nebo jeho aktivita vykazuje nějaký defekt, dochází ke kumulaci lyzozomálního glykogenu v mnoha tkáních. Nejčastěji bývá postiženo právě příčně pruhované svalstvo. U malých dětí je toto postižení patrné také v srdečním svalu.
Nejvíce se dozvíme o této nemoci, když se podíváme na toho, kdo jí propůjčil své jméno. Joannes Cassianus Pompe byl holandský patolog, který se narodil 8. září 1901 v Utrechtu a jehož vědeckou i životní dráhu bohužel ukončila 2. světová válka. Jeho starší bratr Willem Petrus Joseph Pompe (1893–1968) se angažoval v oblasti práva a kriminologie. J. C. Pompe absolvoval Univerzitu v Utrechtu a během té doby měl možnost studovat spousty příznaků onemocnění dnes známého jako Pompeho nemoc. Roku 1932 tyto poznatky publikoval, a tak se jeho jméno dostalo do povědomí mnoha lékařů. Jeho kariéru lékaře přerušila válka, kdy byl povolán do aktivní služby. Jeho účast v protiněmeckém odporu vedla bohužel až k jeho zatčení 15. dubna 1945, kdy byl spolu s dalšími devatenácti spoluvězni zastřelen odplatou za útok holandského odporu na železnici.
Co vlastně dr. Pompe odhalil?
Právě na základě patologických změn srdce následkem enzymového deficitu dr. Pompe diagnostikoval toto onemocnění, přesněji jeho infantilní formu. Mezi další tkáně a soustavy, které mohou být v malé míře postiženy, patří cévní systém, centrální nervová soustava, játra a ledviny. Studie genu odpovědného za Pompeho onemocnění odhalily až 200 různých mutací a polymorfizmů. Díky tomuto faktu je onemocnění přes svůj ojedinělý výskyt značně klinicky variabilní. A to je fakt, který přitěžuje právě diagnostice.
Klasifikace Pompeho nemoci
Od doby, kdy bylo toto onemocnění poprvé popsáno, uplynulo hodně času a také jeho popis prošel jistými změnami. Dnes je z hlediska Mezinárodní klasifikace nemocí (MKN-10) tato nemoc řazena do kategorie E74.0 – Porucha ukládání glykogenu, podle MKN-11 nese označení 5C51. Zde nalezneme několik různých onemocnění řazených pod poruchy ukládání glykogenu. Co je zajímavé, je výčet jmen těchto onemocnění (tabulka 1).
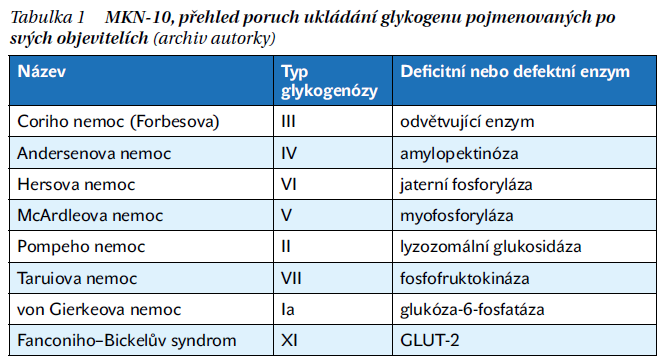
Co se skrývá za jmény objevitelů?
Pokud se podíváme na jmenná označení nemocí, vypadá to trošku, jako by si v této kategorii nemocí dali dostaveníčko lékaři a odborníci z celého světa. Dorothy Hansine Andersen byla Američanka a žena (takže by možná mělo onemocnění znít nemoc Andersenové), Coriho nemoc je pojmenována po manželech Coriových, kteří byli v době pojmenování nemoci již také Američané. Velice zajímavý je fakt, že se Carl Ferdinand Cori narodil 5. 12. 1896 v Praze a jeho žena Gerty Theresa Cori se narodila 15. 8. 1896 taktéž v Praze, oba tedy ještě v době Rakouska-Uherska. Emigrovali v roce 1920 do Ameriky a v roce 1947 obdrželi Nobelovu cenu za medicínu právě za rozsáhlý výzkum na glykogenu. Seiichiro Tarui byl japonský lékař, Edgar Otto Conrad von Gierke a Horst Bickel byli němečtí lékaři, Guido Falconi byl Švýcar, a jak již bylo zmíněno na začátku, Joannes Cassianus Pompe byl holandského původu.
Přes velké množství označení a dělení od roku 1932, kdy Pompe popsal toto onemocnění, se zachovalo pouze základní dělení, a to kategorie infantilní Pompeho nemoc a pozdní Pompeho nemoc. Pozdní forma může být ještě dále členěna na juvenilní a adultní formu. Některá literatura uvádí další dělení infantilní formy na klasickou (do jednoho roku věku) a neklasickou (do dvou let) a dělení pozdní formy na pozdní dětskou a dospělou variantu, jiná slučuje „neklasickou“ formu do dětské varianty progredující do pozdějšího věku pacienta.
Statistika s podivnými výsledky
Pompeho nemoc je velmi vzácné onemocnění. Na druhou stranu jeho povědomost mezi lidmi je minimální a příznaky bývají často buď zcela ignorovány, nebo zaměňovány za příznaky jiných onemocnění (tabulka 2). Přitom data ukazují, že u zvýšené hladiny kreatinkinázy se pravděpodobnost záchytu Pompeho nemoci pohybuje mezi 2–4 % a u pletencové svalové dystrofie je to dokonce 5–15 %.

Vlivem autozomálně recesivně vázané dědičnosti postihuje Pompeho nemoc stejnoměrně obě pohlaví. Incidence onemocnění zahrnující infantilní i pozdní formu se pohybuje statisticky kolem hodnot 1 : 40 000. Klasická infantilní forma je popisována až 1 : 138 000, a naopak pozdní forma 1 : 57 000 (hodnoty z holandských studií). Statistická data ve světě i u nás jsou ovšem velmi neúplná díky malé povědomosti o onemocnění, a zatímco teorie říká, že by mělo být v České republice průměrně kolem 150 pacientů, data z roku 2007 mluvila o pouhých 4 diagnostikovaných pacientech. Interval diagnosticky také není statisticky příliš příznivý. Sledováním v některých zemích Evropy (nejvíce jsou rozšířené vědomosti o této nemoci samozřejmě v Holandsku) bylo zjištěno, že interval mezi prvními příznaky a následnou diagnostikou se pohybuje mezi 7–9 roky. U velmi malých dětí se minimální doba pro diagnostiku pohybuje kolem 3 měsíců, proto se může stát, že u infantilní formy miminko zemře dříve, něž lékaři Pompeho nemoc diagnostikují.
Projevy onemocnění
Popsat projevy vzácného onemocnění není úplně jednoduché. V důsledku malého počtu statistických dat jsou některé formy popsány lépe a některé hůře. Infantilní forma se vlivem rychlé progrese onemocnění snadněji sleduje a také je právě tato forma Pompeho nemoci nejlépe prostudována. Charakteristika je ovšem velmi nemilosrdná.
Klasická infantilní forma postihuje děti v kojeneckém věku, přesně tedy ve věku do jednoho roku. Onemocnění se rozvíjí přibližně v první třetině roku věku dítěte a má poměrně rychlý průběh. V dětském organizmu je nulová aktivita GAA, následkem čehož se extrémně kumuluje glykogen, a to nejvíce v kosterním svalstvu a v srdečním svalu. Nejčastější projevy klasické infantilní Pompeho nemoci jsou tři: muskuloskeletární, respirační a gastrointestinální.
Miminka jsou hypotonická (označováno často jako „flopy infant syndrom“), lze u nich pozorovat zpoždění v tělesném vývoji oproti standardům – zvláště pak motorické schopnosti a zhoršující se svalovou slabost, trpí poruchami příjmu potravy a často lze všechny potíže shrnout do označení všeobecného neprospívání. Mezi další příznaky lze řadit hypertrofickou kardiomyopatii, kardiomegalii, mírnou hepatomegalii a makroglosii. Z respiračních potíží jsou to zvýšená dechová frekvence, oslabení respiračního svalstva, zapojení auxiliárního dýchacího svalstva. S tím souvisí extrémní citlivost vůči infekcím horních cest dýchacích a spánková apnoe. Vlivem rychlé progrese onemocnění takto postižené miminko umírá následkem selhání jak srdečních, tak plicních funkcí, a to do jednoho roku věku.
Mimo klasickou formu infantilní Pompeho nemoci byla popsána i „neklasická“. Zde je ještě jakási měřitelná aktivita enzymu, ale jediný rozdíl oproti variantě klasické je o něco pozdější nástup onemocnění, mírnější progresejednotlivých příznaků, a tím posunuté úmrtí malého pacienta mezi první a druhý rok věku. Otázkou je, co je v případě infantilní formy Pompeho nemoci příznivější, zda klasická, či neklasická forma. Psychický dopad na rodiče vlivem ztráty miminka je v obou případech obrovský.
Diagnostika
U dětské formy se sledují základní obtíže, nástup prvních příznaků, dědičné zatížení (svalové onemocnění v rodině), dále jsou to odborná vyšetření v oblasti neurologie, kardiologie a plicní vyšetření. Typickým příznakem je projev novorozence v tzv. trakčním testu (úchop za horní končetiny a tah jakoby směrem do sedu), zde se projevuje hypotonie, chybí kontrola držení hlavičky. Dalším příznakem je například při kojení špatná koordinace úkonů sání/polykání, výrazná přítomnost pocení a viditelné vyčerpání miminka. Někdy se může objevit až gastroezofageální reflux. Sací a dávivý reflex jsou u dítěte obecně slabší. Mimo sledování svalstva je také nutné sledovat nepřirozenou činnost srdce, jako jsou srdeční šelesty a arytmie.
Z typických vyšetření sem patří EKG a rentgen hrudníku, z laboratorních jsou to testy na jaterní a svalové enzymy a přítomnost oligosacharidů v moči. Dále je to samotné měření aktivity GAA enzymu a hladiny kumulace glykogenu v organizmu.
U pozdních forem onemocnění má progrese velmi pozvolný charakter, což přispívá k delšímu časovému odstupu mezi prvním příznakem a finální diagnostikou. Onemocnění, které se dostavuje až „plíživě“ způsobí, že si jej ani samotní pacienti ani lidé žijící ve společné domácnosti nemusí vůbec povšimnout.
Prvním příznakem je zaostávání ve fyzické aktivitě za vrstevníky. Ve školním věku může být i toto vodítko přehlédnuto, zvláště, pokud bude tělesná konstituce malého pacienta mimo „standard“. U extrémně hubeného dítěte nebo naopak u dítěte s nadváhou se předpokládá jistá neobratnost, takže skutečný pacient nejspíš skončí v prvních fázích onemocnění s dvojkou z tělocviku než na vyšetření pro podezření na vzácné metabolické onemocnění. Nejčastější projevy shrnuje tabulka 3.

Dalším postupem onemocnění je i regrese již získaných motorických schopností, jako je ztížená schopnost zvednout tělo z dřepu či lehu nebo výstup do schodů. Veškerá tělesná námaha je pak provázena zvýšenou dušností. Problematický se stává i spánek pacienta, objevuje se spánková apnoe, jejímž následkem může být až snížená saturace centrální nervové soustavy kyslíkem. Navenek se tento stav projevuje ranními bolestmi hlavy, které se zdají být jakoby bez zjevné příčiny.
Progredující onemocnění následně způsobuje u pacienta neschopnost pohybu bez pomoci druhých, bývá upoután na invalidní vozík. Respirační potíže gradují až do fáze, kdy je pacient odkázán na přístrojovou podporu dechu. Nejčastější příčinou úmrtí pacientů u pozdní fáze Pompeho nemoci je selhání dýchání.
Jak je z již uvedeného zřejmé, i pacient s pozdní formou může na toto onemocnění zemřít. Na začátku jsme se zmínili o tom, že věk hraje určitou roli v budoucnosti a přežití pacienta. Teď je asi ten správný okamžik toto specifikovat. Věk je sice důležitým faktorem z hlediska statistiky úmrtí pacienta, ale vztažen pouze na dělení infantilní a pozdní formy Pompeho nemoci. Zatímco u miminek je statisticky méně pravděpodobné, že malý pacient přežije, u pozdních forem je tato pravděpodobnost značně vyšší. Závisí to samozřejmě ale i na dalších faktorech.
Tím nejdůležitějším je u pozdní formy nikoliv věk, ale pouze a jen typ mutace onemocnění a dále zbytková aktivita enzymu GAA. Ten je rozhodující a k němu se může přidat následně i včasnost diagnostiky a případně léčby. Dnes mají pacienti pozdních forem výrazně větší šanci než třeba před dvaceti lety díky rozvoji diagnostiky, a hlavně novým možnostem v oblasti léčby.
Screeningové vyšetření „suché kapky“
V roce 2007 byli s tímto onemocněním diagnostikování v ČR pouze 4 pacienti. Od roku 2008 probíhal v rámci osvěty spojené s Pompeho nemocí projekt na zvýšení informovanosti veřejnosti, jak odborné, tak laické, spolu se snahou „dodiagnostikovat“ rizikové pacienty a doplnit statistická data v České republice. Primárně se tento projekt týkal neurologických pracovišť pro dětské i pro dospělé pacienty, kde se předpokládal nejvyšší výskyt potenciálně nemocných. Projekt probíhal za spolupráce fakultních brněnských neurologických pracovišť a univerzitní kliniky v Hamburku za podpory společnosti Genzyme (v současnosti Sanofi).
Pokud bychom porovnali počty evidovaných pacientů například s Belgií, která má, ač menší rozlohou přibližně stejný počet obyvatel, pak v roce 2008 to bylo na 5 českých pacientů 38 těch belgických. Je otázkou, zda je to dáno větší četností onemocnění, nebo tím, že má Belgie k Holandsku blíže.
Jednoduché vyšetření enzymatické aktivity pomocí metody „suché krevní kapky“ (dried blood spot – DBS) je nenáročné a efektivní jak z hlediska pacienta samotného, tak z hlediska lékařů a i nákladů zdravotních pojišťoven. Pokud je test suché kapky pozitivní, je vyšetření doplněno o další enzymologické vyšetření, případně o genetické vyšetření pro stanovení mutace genu. Právě rozmanitost genových mutací ztěžuje primární diagnostiku a typ mutace pak výrazně ovlivňuje průběh samotného onemocnění.
Co je ovšem velmi zajímavé, je fakt, že tento typ screeningu se provádí i u novorozenců k odhalení některých onemocnění. Testování se provádí 48–72 hodin po narození dítěte odběrem krve z patičky dítěte na speciální diagnostický papírek. Od roku 2009 bylo standardně zařazeno 13 onemocnění, která se takto testují, od června 2016 je to 18 onemocnění. Bohužel Pompeho nemoc v nich zatím zahrnuta není.
Léčba
Pompeho onemocnění bylo ještě před 10 lety jen velice obtížně léčitelné, a pacienti s pozdní formou tak mohli jen sledovat negativně probíhající postup onemocnění. Na konci 20. století vznikly projekty na výzkum možné substituční léčby tohoto onemocnění a první preparát byl připraven ke klinickým studiím v roce 1999. V roce 2007 byl v celé Evropě registrován preparát Myozyme, který nabízí vhodnou enzymatickou substituční léčbu (, enzyme replacement therapy – ERT) ve formě intravenózního podání. Substituční léčba je dávkována podle váhy pacienta.
Substituční enzymatická léčba je možností nejen pro pozdní formu, u dospělých může až zcela zastavit progresi onemocnění, minimálně ale dochází ke zpomalení příznaků a k oddálení potřeby invalidního vozíku a umělé plicní ventilace.
Statistická data ovšem ukazují, že i u dvouletých dětí došlo k výraznému zlepšení a enzymoterapie může vést až vyloučení úmrtí u infantilní formy. Důležité je ovšem včasné odhalení nemoci a okamžité zahájení léčby. Výzkumy v oblasti Pompeho nemoci stále pokračují a doufejme, že výsledky přinesou pro pacienty další výhody.
Substituční terapie ovšem není jedinou léčbou Pompeho nemoci. Standardně musí být (jako už u všech potíží pohybového aparátu) kombinována s rehabilitací, posilováním svalového korzetu, balneoterapií a další symptomatickou léčbou.
Vzácná onemocnění obecně
Vzácná onemocnění jsou nejčastěji definována jako počet evidovaných onemocnění na 10 000 obyvatel. Podle evropské legislativy je jako „vzácné“ označeno onemocnění s prevalencí 5 a méně postižených na 10 000 osob.
Podchycení vzácných onemocnění, jejich evidence, sběr dat a díky tomu také lepší finální péče pro pacienty jsou jednoznačným cílem webového portálu Orphanet, který byl v české verzi spuštěn v červenci 2021 jako 8. jazyková mutace.
Závěr
Pompeho nemoc je riziková ve své klinické variabilitě a nízké pravděpodobnosti, že se s ní běžný praktický lékař u svého pacienta setká. Vzácným onemocněním, jako je glykogenóza, pomůže jen to, když se informace o jejich existenci bude šířit mezi zdravotníky i pacienty všemi možnými a dostupnými prostředky. Čím více toho víme a čím více se snažíme si nastudovat, tím více zjišťujeme, že naše znalosti jsou pouhým střípkem v obrovské skládačce. To, co je ovšem důležité, je nepřestat hledat a doplňovat další dílky informací do finálního obrazu. Buďme vděčni takovým lékařům, jako byl doktor Pompe, kteří se nespokojí s jednoduchými vysvětleními a hledají odpovědi i u nepatrných odchylek od normálu.
RNDr. Lenka Grycová