Vaskulitidy se zpravidla dělí podle velikosti postižených cév na vaskulitidy velkých, středních a malých cév. Diagnostika se opírá o klinický obraz, laboratorní a histologické nálezy, včetně zobrazovacích metod a vyžaduje nezbytnou mezioborovou spolupráci. Nové poznatky o patogenezi vaskulitid přispěly k využití moderních terapeutických postupů, včetně biologické terapie. V tomto přehledovém sdělení se zaměřujeme na problematiku vybraných primárních vaskulitid, konkrétně na obrovskobuněčnou arteriitidu, polyarteritis nodosa a vaskulitidy asociované s protilátkami proti cytoplazmě neutrofilních leukocytů.
Vaskulitidy představují heterogenní skupinu onemocnění, která často provází variabilní kožní projevy a systémový charakter s celkovými příznaky. V případech, kdy jsou způsobeny nežádoucími účinky léků, následkem infekčních onemocnění, malignitami nebo autoimunitními chorobami, hovoříme o sekundárních vaskulitidách. Pokud je příčina neznámá, jedná se o primární vaskulitidy, jejichž výskyt je velmi vzácný.
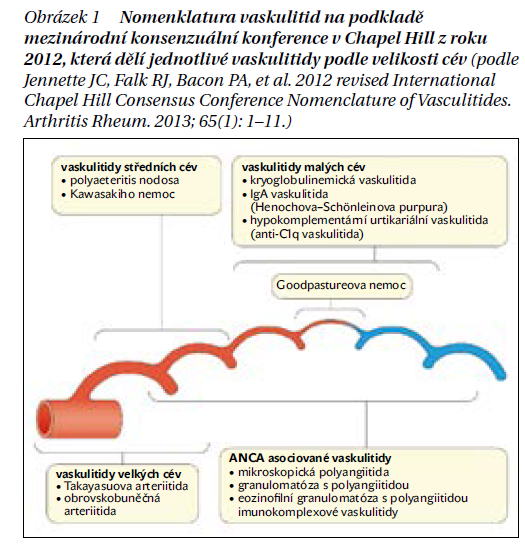
Nomenklatura vaskulitid je založena na mezinárodní konsenzuální konferenci v Chapel Hill a poskytuje klíčový rámec pro klasifikaci těchto onemocnění, což usnadňuje rozlišení mezi jednotlivými formami (obrázek 1). Vaskulitidy se dělí podle převažující velikosti postižených cév na ty, které postihují cévy velkého, středního a malého kalibru. Při podezření na vaskulitidu je nezbytné určit rozsah postižení, vyloučit možné vaskulitické mimikry a potvrdit diagnózu. Klinická prezentace, výsledky laboratorních testů, zobrazovací metody a často i bioptický nález jsou pro diagnostiku klíčové.
Primární vaskulitidy jsou charakterizovány multiorgánovým postižením, které vzniká v důsledku zánětu a fibrinoidní nekrózy cévní stěny. To vede k poškození integrity cévního endotelu, krvácení, mikrotrombotizaci a postupné obliteraci cévního lumen, což přispívá k ischemii postižené tkáně a rozmanitému klinickému obrazu. Na vývoji vaskulitid se podílí různé imunopatologické mechanismy, zahrnující zánětlivé buňky, ukládání imunitních komplexů nebo protilátek do cévní stěny.
Klinický obraz vaskulitid je rozmanitý. Většina pacientů má celkové příznaky systémového zánětlivého onemocnění, jako jsou horečka, únava, celková slabost, ztráta chuti k jídlu a hubnutí. Mezi další časté příznaky patří bolesti kloubů a svalů, kožní změny, dýchací potíže a neurologické problémy. Specifické symptomy se liší v závislosti na konkrétním typu onemocnění a postižených orgánech, což je ovlivněno velikostí postižených cév.
V tomto přehledovém sdělení se zaměříme na diagnostiku a léčbu vybraných primárních vaskulitid, konkrétně na obrovskobuněčnou arteritidu a nekrotizující systémové vaskutlitidy typu polyarteritis nodosa a vaskulitid asociovaných s protilátkami proti cytoplazmě neutrofilních leukocytů (ANCA).
Obrovskobuněčná arteriitida
Obrovskobuněčná (Hortonova) arteriitida je charakterizována jako granulomatózní zánět velkých a středních cév. Tradičně byla popisována jako temporální arteriitida, ale v současnosti je chápána jako systémové onemocnění zahrnující aortu a všechny její hlavní odstupující větve. Patří mezi nejčastější systémové vaskulitidy s roční incidencí kolem 10–40 případů na 1 milion obyvatel starších 50 let s prevalencí kolem 200 případů na 1 milion obyvatel starších 50 let. Častěji postihuje ženy a vrchol výskytu se pohybuje mezi 70.–80. rokem věku.
Existují 3 základní podtypy onemocnění:
- klasická neboli čistě kraniální temporální arteriitida;
- temporální arteriitida s postižením velkých cév;
- izolované extrakraniální onemocnění velkých cév bez kraniálních příznaků.
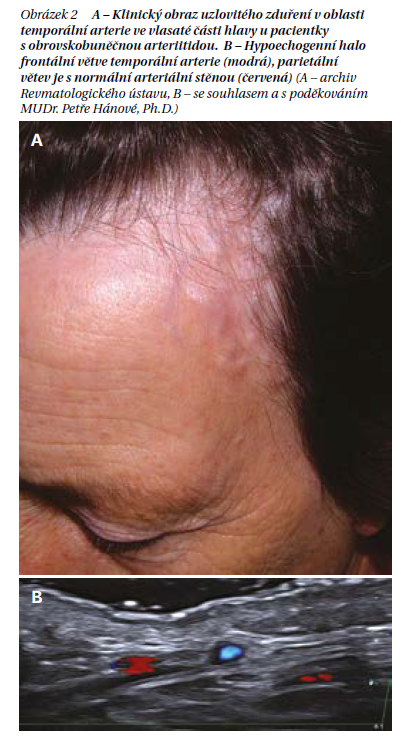
Mezi typické kraniální příznaky patří bolesti hlavy, citlivost kštice, klaudikace čelisti a jazyka nebo křeč žvýkacích svalů, nebo poruchy zraku, které patří mezi specifické ischemické komplikace. Při fyzikálním vyšetření lze odhalit uzlovité zduření a palpační bolest ve vlasaté části hlavy (obrázek 2), zejména v oblasti temporální arterie, mohou být i oslabené pulzace.
Varovné příznaky katastrofického scénáře zahrnují neostré vidění, výpadky zorného pole, diplopii a zejména přechodnou ztrátu zraku (amaurosis fugax), která může vést k trvalému oslepnutí. Oslepnutí je často náhlé a nebolestivé a je důsledkem vaskulitidy zadních ciliárních větví oftalmické tepny, což vede k ischemickému postižení optického nervu, známému jako AION (anterior ischemic optic neuropathy). Při výskytu varovných příznaků by nikdy neměla být terapie odkládána.
Časté jsou nespecifické celkové příznaky, jako je nechutenství, hubnutí, febrilie a zvýšená únavnost. V přibližně polovině případů se objevují symptomy revmatické polymyalgie, které se projevují postupně se rozvíjejícími silnými bolestmi v obou ramenou, krku, šíji a pánevním pletenci, s maximální intenzitou ráno, a někdy také bolestmi kloubů, případně artritidou. V závislosti na postižené tepně mohou pacienti zažívat tinitus, ztrátu sluchu, závratě, tranzitorní ischemickou ataku nebo dokonce mozkovou příhodu. Dalšími možnými projevy jsou klaudikace horních končetin, kašel, bolesti břicha nebo vážné příznaky spojené s aneuryzmatem nebo disekcí aorty. Při postižení aorty a jejích odstupujících větví může být pozorována rozdílnost krevního tlaku a pulzace v horních končetinách, případně šelest v oblasti karotidy nebo podklíčkové tepny. Při těchto příznacích je nutné uvažovat o Takayasuově arteriitidě, která má přibližnou incidenci kolem 1 případu na 1 milion obyvatel. Na rozdíl od obrovskobuněčné arteriitidy postihuje mladé jedince kolem 25 roku věku, obvykle ženy. Diagnosticky se nejlépe uplatňuje magnetická rezonance (MR) s angiografií.
Při izolovaném postižení aorty je důležité vyloučit bakteriální příčinu. Klinický obraz izolované neinfekční aortitidy zpravidla odpovídá obrovskobuněčné arteriitidě bez kraniálních příznaků. Často se jedná o projev IgG4 asociovaného onemocnění. V některých případech (až v 10 %) je izolovaná aortitida diagnostikována náhodně během chirurgického zákroku na aneuryzmatu aorty. Nález chronické periaortitidy s postižením břišní aorty může odpovídat idiopatické retroperitoneální fibróze; někdy se objevuje zánětlivá abdominální aortitida bez přítomnosti aneuryzmatu či retroperitoneální fibrózy.
Laboratorně je patrná zvýšená hladina reaktantů zánětu, často dosahující stovkových hodnot sedimentace erytrocytů a C-reaktivního proteinu (CRP). Většina pacientů trpí anémií chronických onemocnění. Biopsie temporální arterie se provádí v nejasných případech a může odhalit granulomatózní zánět, mononukleární a lymfocytární infiltraci s fragmentací lamina elastica interna. Mnohojaderné obrovské buňky se prokáží asi v polovině případů.
V současnosti jsou při diagnostice preferovány zobrazovací metody, jako první se doporučuje sonografické vyšetření temporální a axilární arterie, které odhalí transmurální zánět cévní stěny, tzv. halo příznak (obrázek 3). V další fázi je možné využít MR s vysokým rozlišením nebo pozitronovou emisní tomografii s využitím deoxyglukózy značené radioaktivním fluorem (FDG-PET). Konvenční angiografie se pro diagnostiku obrovskobuněčné arteriitidy nedoporučuje. Pro monitorování aktivity onemocnění obvykle postačuje klinické vyšetření a sledování hladin reaktantů akutní fáze.
Základem indukční léčby jsou glukokortikoidy ve vysoké dávce, obvykle se podává 40–60 mg prednisonu nebo jeho ekvivalent denně. Při varovných příznacích očního postižení, ale i při již navozené ztrátě zraku, je třeba bez prodlení zahájit pulzní léčbu methylprednisolonem 1 g denně po dobu 3 dnů a poté zahájit prednison 60 mg denně s pozvolnou redukcí dávky podle klinického stavu a poklesu reaktantů zánětu. Účinek lze obvykle pozorovat během několika málo dnů. Po 3 měsících léčby by se mělo dosáhnout dávky 15–20 mg prednisonu denně. Léčba většinou trvá kolem 2 let.
Monoterapii glukokortikoidy lze považovat za možnost udržení remise v případech, kdy se po 1 roce podaří dosáhnout cílové dávky ≤ 5 mg prednisonu denně. U více než poloviny pacientů je však nezbytné přidat methotrexát.
Schválenou indikací pro refrakterní a relabující formu onemocnění je tocilizumab, biologický lék zasahující na úrovni interleukinu 6 (IL-6). V úvodu se doporučuje nasadit inhibitory protonové pumpy a preventivní opatření proti osteoporóze, včetně její případné léčby. Rutinní nasazení anopyrinu se již nedoporučuje, pokud k tomu není jiná indikace. Prognóza pacientů je většinou dobrá, přičemž největší komplikací je ztráta zraku, která stále postihuje kolem 15 % případů. Revaskularizace se u obrovskobuněčné arteriitidy provádí zřídka, častěji však u pacientů s Takayasuovou arteriitidou; někdy je třeba provést operaci aneuryzmatu.
Polyarteriitis nodosa
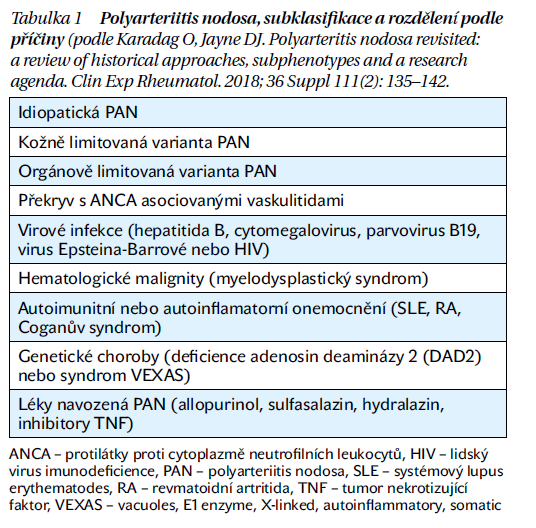
Polyarteriitis nodosa (PAN) je vzácné multisystémové onemocnění charakterizované nekrotizujícím zánětem středně velkých a menších cév periferních nervů a kůže, méně často gastrointestinálního traktu, ledvin, svalů a vzácně i dalších orgánů. Prevalence nepřesahuje 30 pacientů na 1 milion obyvatel. Ženy jsou pravděpodobně postiženy o něco častěji než muži, onemocnění se manifestuje kolem 40.–60. roku věku. Rozeznáváme primární a sekundární formu PAN (tabulka 1). V minulosti se rozeznávala PAN idiopatická a asociovaná s hepatitidou B, současné spektrum sekundární PAN se rozšířilo o stavy asociované s infekcemi, paraneoplastické projevy nebo nově popsané autoinflamatorní onemocnění, včetně genetických syndromů.
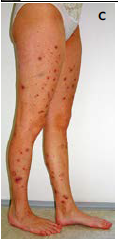
Průběh onemocnění je variabilní, ve většině případů jsou celkové projevy se zvýšenými teplotami, hubnutím, bolestmi kloubů a svalů. V klinickém obrazu dominuje u více než dvou třetin pacientů bolestivá asymetrická senzomotorická periferní neuropatie. Postižení kraniálních nervů je vzácné. V polovině případů je přítomna purpura, noduly, livedo retikularis nebo nekrotické ulcerace. Postižení gastrointestinálního traktu je spjato se špatnou prognózou, mortalita se pohybuje kolem 25 %. Mezenteriální vaskulitida se projevuje bolestí břicha, může vést k intestinální ischemii, perforaci a krvácení. Při postižení ledvin je hlavním projevem hypertenze, mikroskopická hematurie a malá proteinurie, glomerulonefritida nebývá přítomna. Postižení srdce je vzácné, vaskulitida koronárních arterií může navodit infarkt myokardu a na podkladě hypertenze může nastat hypertrofická kardiomyopatie a srdeční selhání. Méně častá je bolest varlat při orchitidě.
Diagnostika PAN je těžká, protože se onemocnění manifestuje nespecifickými a heterogenními projevy. Diagnostický proces je založen na klinickém obrazu a bioptickém vyšetření postiženého orgánu. V případě pozitivního elektromyografického nálezu je doporučena biopsie nervus suralis. Provádí se také biopsie kůže, svalu, rekta nebo testes. Biopsie ledvin není často výtěžná. V případě nemožnosti provést biopsii postiženého orgánu se doporučuje angiografie.
Charakteristickými histologickými nálezy jsou fokálně segmentální vaskulitida cév středního a menšího kalibru s transmurální zánětlivou infiltrací cévní stěny polymorfonukleárními leukocyty, fibrinoidní nekrózou a přítomností chronických fibrotických změn, které přispívají ke vzniku aneuryzmat a stenóz. Laboratorní testy jsou obvykle nespecifické, lze pozorovat leukocytózu, trombocytózu a zvýšení hladin reaktantů akutní fáze. Autoprotilátky jsou zpravidla negativní. U menšího procenta pacientů může být pozitivní HBsAg. U mladších jedinců je třeba zvážit možnost autozomálně recesivní imunodeficience s mutací v genu pro adenosin deaminázu 2.
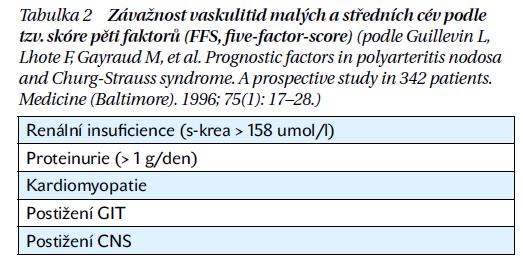
Léčba PAN se řídí závažností onemocnění podle tzv. skóre pěti faktorů (FFS), podobně jako u vaskulitid cév menšího kalibru (tabulka 2). Základem terapie je indukční léčba, následovaná udržovací terapií a léčbou při relapsu onemocnění. V případě mírné formy onemocnění, jako jsou celkové projevy, muskuloskeletální postižení a kožní projevy, je možné podat kortikoterapii v monoterapii.
Při refrakterním průběhu se může zvážit kombinace s methotrexátem nebo azathioprinem. U pacientů s limitovanou formou pouze na kůži obvykle není nutná imunosupresivní léčba. V případě vážného průběhu onemocnění (FFS ≥ 1), který ohrožuje funkci orgánů nebo život pacienta, je doporučena vysokodávková kortikoterapie s intravenózními pulzy methylprednisolonu v dávce 0,5–1 g po dobu 3 dnů, kombinovaná s cyklofosfamidem v pulzech odpovídajících zhruba 10 mg/kg tělesné hmotnosti po 3 týdnech, s maximálně šesti pulzy. Udržovací léčba zahrnuje methotrexát nebo azathioprin po dobu 18 měsíců. Při relapsu existují pozitivní výsledky s biologickou léčbou (tocilizumab, inhibitory TNF nebo rituximab), zvážit je možné intravenózní imunoglobuliny, inhibitory Janusových kináz nebo plazmaferézu.
Při sekundární PAN je nezbytné léčit základní příčinu, jako je hepatitida nebo myelodysplastický syndrom. U vaskulitidy spojené s deficitem adenosin deaminázy 2 jsou účinné inhibitory TNF.
Granulomatóza s polyangiitidou
Granulomatóza s polyangiitidou (GPA), dříve známá jako Wegenerova granulomatóza, představuje primární systémovou vaskulitidu, která je charakterizována nekrotizujícím zánětem cévních stěn a tvorbou granulomů. Typické postižení se projevuje v oblasti dýchacích cest a ledvin. Jedná se o velmi vzácné onemocnění, jehož roční incidence je až 10 případů na 1 milion obyvatel. Postižení žen a mužů je přibližně stejné a typický věk pro nástup onemocnění se pohybuje mezi 45 a 65 lety.
GPA má heterogenní průběh. V případě klasické generalizované formy je postižen jak horní, tak dolní respirační trakt i ledviny, a naopak při limitované GPA nejsou ledviny postiženy a v popředí je infiltrace granulomy mimo cévy bez projevů vaskulitidy, např. sinusitida, otitis media nebo oční obtíže.
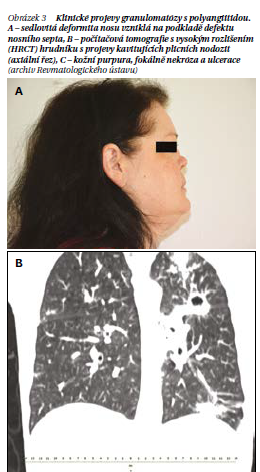
Horní dýchací cesty vykazují chronický zánět v podobě purulentní rýmy a sinusitidy, pocitu ucpaného nosu a epistaxe, slizničních ulcerací a méně často defektů nosního septa, které mohou vést k rozvoji sedlovité deformity nosu (obrázek 3A). Obstrukce Eustachovy trubice přispívá k recidivujícím zánětům středouší, což by u dospělých jedinců mělo vyvolat podezření na GPA. Postižení laryngu a trachey se projevuje chrapotem a subglotickou stenózou. Až 90 % pacientů trpí postižením dolních dýchacích cest, které může mít variabilní průběh. Někdy může být asymptomatický, častý však je kašel, může docházet k hemoptýze a bolesti na hrudi. Akutní kapilaritida a zánětlivá infiltrace intersticia a alveolů mohou vést až k život ohrožující alveolární hemoragii. Chronické zánětlivé změny se projevují tvorbou granulomů a nodulů na plicích (obrázek 3B). Výsledkem postižení dýchacích cest může být chronická respirační insuficience. Intersticiální plicní onemocnění jsou vzácná, častější jsou respirační infekce způsobené obstrukcemi dýchacích cest (např. bakteriální sinusitidy, pneumonie) nebo jako komplikace imunosupresivní léčby (Pneumocystis jirovecii).
Postižení ledvin v úvodu nemoci není běžné a často je asymptomatické, což může vést k přehlédnutí. Typicky se projevuje mikroskopickou hematurií, přítomností erytrocytárních válců v močovém sedimentu a občasnou proteinurií. Neléčené postižení ledvin může vést k renálnímu selhání. Ledviny jsou během nemoci postiženy přibližně u dvou třetin pacientů. Histologicky se jedná o pauciimunní rychle progredující nekrotizující glomerulonefritidu se srpky.
Celkové příznaky jsou u pacientů s generalizovanou formou GPA běžné. Myalgie a artralgie se vyskytují u dvou třetin pacientů. Oční postižení je přítomno přibližně u poloviny případů a může být různorodé, zahrnující širokou škálu očních struktur; často může být i počátečním projevem onemocnění. Útlak optického nervu může vést ke ztrátě zrakové ostrosti, může být přítomna skleritida, episkleritida atd. Exoftalmus je vzácný, projevuje se granulomatózním zánětem v orbitě. Postižení vnitřního ucha se projevuje ztrátou sluchu a vertigem. Na kůži se u asi poloviny pacientů objevuje hmatná purpura, podkožní noduly, ulcerace nebo livedo reticularis. Menší část pacientů může trpět periferní neuropatií (mononeuritis multiplex, symetrická polyneuropatie) a v některých případech může být postižen i centrální nervový systém (pachymeningitida, mozkové krvácení, křeče atd.). Granulomatózní postižení dalších orgánů, včetně srdce, je vzácné.
V krevním obrazu dominuje leukocytóza a trombocytóza doprovázená obvykle zvýšením hladiny reaktantů akutní fáze. Téměř všichni pacienti mají pozitivní ANCA protilátky, naprosto převažuje cytoplazmatický typ fluorescence, specifické jsou PR3-ANCA protilátky, které jsou přítomny až u 90 % pacientů s generalizovanou formou GPA, zatímco u limitované formy jsou méně časté (asi 60 %). ANCA obvykle korelují s aktivitou onemocnění. Zobrazovací metody, zejména CT a MR, jsou často využívány k diagnostice, stejně jako biopsie postižených orgánů, přičemž ledviny jsou nejčastěji vyšetřovaným orgánem. Mezioborová spolupráce je nesmírně důležitá pro efektivní diagnostiku. Diagnóza GPA je založena na klinickém obrazu, sérologických nálezech, zobrazovacích metodách a často i histologických vyšetřeních. Průměrné zpoždění od prvních příznaků k definitivní diagnóze obvykle činí kolem 6 měsíců. U limitované formy onemocnění může být toto zpoždění ještě delší, často z důvodu nižší prevalence ANCA protilátek a menší diagnostické výtěžnosti biopsie horních cest dýchacích.
Léčba GPA zahrnuje indukční a udržovací terapii. Pro dosažení remise u pacientů se závažným orgánovým postižením se doporučuje kombinace vysokých dávek glukokortikoidů (prednison v dávce 50–75 mg denně) spolu s rituximabem nebo s pulzy cyklofosfamidu. Při těžké renální insuficienci a difuzním alveolárním krvácení je běžnou praxí podávat na začátku pulzní léčbu methylprednisonolem v dávce 1 g po dobu 3 dnů. Postupná redukce prednisonu podle protokolu na denní dávku 5 mg během 4–5 měsíců je důležitá pro minimalizaci vedlejších účinků. K dosažení nízké dávky prednisonu je možné po dobu prvního roku zvážit přidání léku avakopan, což je perorální inhibitor receptoru komplementové složky C5a. Rutinní plazmaferéza se obvykle nedoporučuje, ale je možné ji zvážit u pacientů s renální insuficiencí a rychle progredující glomerulonefritidou. V některých případech je možné zvážit přidání intravenózních imunoglobulinů.
Pro udržení remise se často doporučuje podávání rituximabu, v případě jeho kontraindikace je možné zvážit azathioprin nebo methotrexát. Udržovací léčba obvykle trvá 2–4 roky. Během prvního roku léčby jsou časté infekční komplikace. Proto se rutinně doporučuje profylaxe pneumocystové pneumonie podáváním kombinace trimethoprimu se sulfamethoxazolem, což může snížit i riziko dalších bakteriálních infekcí přispívajících k relapsu GPA. Díky pokroku v léčbě se v posledních desetiletích výrazně snižuje mortalita u pacientů s GPA, pětileté přežití se pohybuje kolem 70–80 %.
Mikroskopická polyangiitida
Mikroskopická polyangiitida (MPA) představuje systémovou nekrotizující vaskulitidu malých až středně velkých cév, která postihuje nejčastěji ledviny a méně často plíce, kůži a další orgány. Na rozdíl od PAN není pro MPA charakteristický vznik cévních mikroaneuryzmat a rovněž se odlišuje od GPA tím, že nedochází ke tvorbě granulomů. Výskyt MPA je nepatrně méně častý než GPA, postihuje mírně častěji muže a obvykle se začíná projevovat mezi 30 a 50 lety věku.
Onemocnění vykazuje značný překryv s GPA. Téměř všichni pacienti mají vaskulitidu ledvin, zatímco plíce jsou postiženy asi v polovině případů. Průběh onemocnění může být buď pomalu progresivní s rozsáhlou sklerotizací glomerulů, nebo fulminantní s projevy rychle progredující glomerulonefritidy. Pacienti s MPA často již při diagnóze trpí pokročilým postižením ledvin a mají vyšší pravděpodobnost přechodu do terminálního stadia renální insuficience. Na rozdíl od GPA je u pacientů s MPA častější výskyt obvyklé intersticiální pneumonie; prvním příznakem může být hemoptýza a někdy i difuzní alveolární krvácení. Závažným je výskyt pulmorenálního syndromu. Postižení horního dýchacího traktu je méně časté. Třetinu až čtvrtinu případů MPA provází artralgie, myalgie, periferní neuropatie a kožní léze charakteru purpury až nekrotizujících defektů. Menší část pacientů může mít serózní rinitidu nebo gastrointestinální projevy.
V laboratorních testech nacházíme zvýšené reaktanty akutní fáze, leukocytózu a anémii. Většina pacientů má pozitivitu ANCA (90 %), charakteristické jsou MPO-ANCA (60 %). Pacienti mohou mít asymptomatickou hematurii a malou proteinurii, vzestup sérového kreatininu až po zjevné selhání ledvin.
Diagnóza se opírá o klinický obraz a bioptický průkaz nekrotizující glomerulonefritidy se srpky, bez imunitních depozit a bez granulomů. Postižení dalších orgánů přispívá k diagnostice.
Léčba MPA obvykle následuje strategii podobnou jako u GPA, zahrnující jak indukční, tak udržovací terapii. V případě těžkého postižení ledvin může být indikována plazmaferéza nebo intravenózní imunoglobuliny. Relapsy se vyskytují ve třetině případů. Pětileté přežití je podobné jako u GPA, prognóza je vážná v případě difuzního alveolárního krvácení.
Eozinofilní granulomatóza s polyangiitidou
Eozinofilní granulomatóza s polyangiitidou (EGPA, dříve známá jako syndrom Churga–Straussové), je multisystémové onemocnění s granulomatózním zánětem malých a středních cév. Onemocnění se typicky projevuje pozdním vznikem asthma bronchiale nebo rinosinusitidy s nosními polypy, doprovázené periferní a tkáňovou eozinofilií s nekrotizující vaskulitidou. Nejčastěji jsou postiženy kůže, periferní nervy a plíce. EGPA je nejvzácnější ANCA asociovaná vaskulitida s roční incidencí 0,5–4 případy na 1 milion obyvatel a prevalencí 10–14 případů na 1 milion obyvatel. Výskyt onemocnění se zdá být mezi muži a ženami podobný a obvyklý věk nástupu se pohybuje mezi 49 a 59 lety.
Různé patogenetické mechanismy stojí za heterogenním průběhem s variabilním klinickým fenotypem onemocnění. Identifikujeme tři základní fáze EGPA, které se mohou překrývat (tabulka 3).

- Prodromální fáze obvykle trvá několik let a je charakterizována alergickými projevy, jako je asthma bronchiale a chronická rekurentní rinosinusitida s nosními polypy.
- Periferní a tkáňová eozinofilie může přetrvávat měsíce až roky a projevuje se například chronickou eozinofilní pneumonií, eozinofilní gastroenteritidou s bolestí břicha a krvavými průjmy nebo akutní eozinofilní myokarditidou. Artralgie a myalgie jsou přítomny u poloviny případů. V obou fázích jsou časté rekurentní horečky a zvýšené riziko trombotických komplikací.
- Ve vaskulitické fázi jsou typické celkové projevy, často s ústupem asthma bronchiale. Dominantními příznaky jsou fokálně segmentální glomerulonefritida, periferní neuropatie a purpura. Může se také objevit chronická zánětlivá kardiomyopatie nebo postižení gastrointestinálního traktu s rizikem perforace střev, peritonitidy, pankreatitidy, nebo vzácně i mezenteriální vaskulitidy.
V krevním obraze často pozorujeme anémii a zvýšené hladiny reaktantů akutní fáze, spolu s elevací sérových hladin IgE. Většina pacientů má nespecificky pozitivní revmatoidní faktory. V periferní krvi často nalezneme výrazně zvýšený počet eozinofilů, překračující hodnotu 1,5 × 109/L. ANCA protilátky jsou pozitivní jen u 30–40 % pacientů, převažující je perinukleární typ fluorescence, obvykle se jedná o protilátky proti MPO. Vaskulitické příznaky, jako je glomerulonefritida, periferní neuropatie a purpura, jsou častější u pacientů s pozitivitou ANCA protilátek. Naopak eozinofilní příznaky, například postižení srdce a gastroenteritida, jsou častější u ANCA negativních pacientů. U většiny pacientů se oba fenotypy onemocnění překrývají.
Pro diagnostiku a hodnocení rozsahu postižení jsou užitečné rtg. a HRCT vyšetření plic, kde můžeme pozorovat periferně uložené skvrnité a migrující infiltráty a někdy i opacity typu mléčného skla, malé noduly nebo ztluštění bronchiální stěny. Difuzní alveolární krvácení je vzácné. V některých případech může být nutná bronchoskopie. Spirometrické vyšetření je běžné pro zhodnocení závažnosti asthma bronchiale. Biopsie kůže a nervus suralis mohou být důležité pro potvrzení diagnózy.
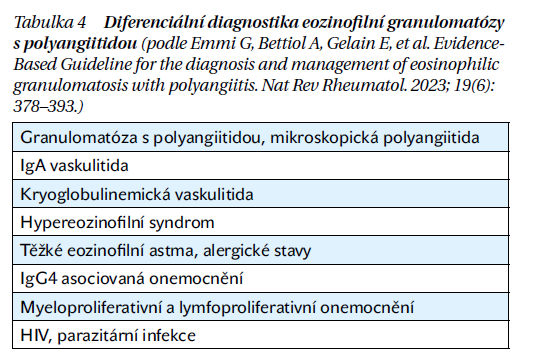
EGPA může mít u každého jedince různý průběh a často je obtížné ji správně diagnostikovat. Klíčovým krokem při stanovení diagnózy je anamnéza asthma bronchiale nebo alergické rinosinusitidy s polypy, přítomnost periferní hypereozinofilie a systémových projevů zánětu s postižením různých orgánů. Bioptické vyšetření odhaluje nekrotizující vaskulitidu a extravaskulární granulomy s centrálně uloženými eozinofily, obklopenými makrofágy a epiteloidními obrovskými buňkami. Vždy je důležité vyloučit jiné hypereozinofilní stavy a další ANCA asociované vaskulitidy (tabulka 4).
Léčba EGPA se řídí závažností onemocnění podle FFS, podobně jako u ostatních ANCA asociovaných vaskulitid. V případě závažného, život ohrožujícího onemocnění s postižením orgánů se podávají pulzy cyklofosfamidu nebo rituximab v kombinaci s intravenózními pulzy methylprednisolonu a následně vysoké dávky perorálních glukokortikoidů s postupnou redukcí dávky.
V případech, kdy není onemocnění tak závažné, lze pro navození nebo udržení remise použít biologickou léčbu zasahující na úrovni IL-5 (mepolizumab nebo benralizumab), dále rituximab nebo konvenční imunosupresivní léčbu, jako azathioprin či methotrexát. Léčba relapsu onemocnění záleží na závažnosti.
Relapsy onemocnění jsou poměrně časté a vyskytují se u více než 60 % pacientů. Prognóza je obecně příznivá; u nekomplikovaných případů přesahuje pětileté přežití 95 %. Riziko úmrtí stoupá s věkem pacienta a počtem postižených orgánů. Nejčastějšími příčinami úmrtí jsou infarkt myokardu a městnavé srdeční selhání.
Závěr
Poslední léta přinesla do diagnostiky a léčby systémových vaskulitid zásadní pokrok. U obrovskobuněčné arteriitidy je nyní silně doporučováno při diagnostice sonografické vyšetření temporální a axilární arterie. Vedle standardních léčebných možností jsou k dispozici biologické léky, které zasahují na úrovni IL-6, a další inovativní terapeutické možnosti prochází pokročilými fázemi klinického hodnocení. Významný pokrok nastal také v diagnostice a léčbě ANCA asociovaných vaskulitid, kde je klíčový důraz kladen na včasnou iniciaci léčby, zlepšení indukční i udržovací fáze remise a redukci celkové dávky glukokortikoidů. To umožňuje několik nových léčivých přípravků. Pro GPA a MPA se již delší dobu využívá úspěšně rituximab a nedávno byl schválen pro indukční fázi léčby inhibitor receptoru komplementové složky C5a avakopan. Nově jsou pro léčbu EGPA, zejména v nezávažných případech, doporučovány biologické léky mepolizumab a benralizumab, které ovlivňují účinek IL-5. S ohledem na vzácný výskyt a heterogenitu těchto onemocnění, jejichž klinické projevy jsou rozmanité a diferenciální diagnóza široká, je klíčové na tato onemocnění pomýšlet a včas je odkazovat k potvrzení diagnózy a zahájení léčby do specializovaných center.
Literatura u autora.
Podpořeno projektem Ministerstva zdravotnictví́ pro koncepční́ rozvoj výzkumné́ organizace 023728.
prof. MUDr. Ladislav Šenolt, Ph.D.
Revmatologický ústav
Revmatologická klinika 1. LF UK, Praha