Spinální muskulární atrofie (SMA) jsou skupinou neurodegenerativních onemocnění motoneuronů nacházejících se v předních rozích míšních a i hlavových nervů. Naprostá většina z nich je způsobena mutací v genu SMN1 (survival motor neuron). Klinicky se projevují podle typu onemocnění různě závažnou progredující svalovou slabostí. Jedná se o vzácné onemocnění, kdy se ročně v České republice narodí přibližně 10 takto nemocných dětí. Jejich prognóza byla (podle typu nemoci) donedávna často vážná, někdy až fatální. Situace se ale změnila v roce 2016. Aktuálně jsou pro léčbu SMA registrována dokonce 3 léčiva fungující na principu genové léčby.
Ani léky na principu genové léčby ale nedovedou obnovit již ztracené motoneurony, a proto je pro dobrou prognózu pacienta nezbytné zahájit jeho léčbu co nejdříve. Léčba již symptomatických pacientů zabrání především progresi nemoci či jen mírně zlepší motorické funkce. Přesto se jedná o významný milník nejen v léčbě neurologických onemocnění, ale o přelom v celé medicíně, protože v případě jednoho z těchto tří léčiv, léku Zolgensma, se jedná o vůbec první systémovou genovou terapii. Pokud je lék podán v presymptomatickém stadiu, je šance, že se dítě bude vyvíjet jako zcela zdravé.
Genetická podmíněnost SMA
Progresivní úbytek motorických neuronů u SMA vede ke svalové slabosti, která je podle typu onemocnění různě závažná a různě rychle progreduje. Svalová slabost je především proximální a vede k pohybovým potížím. Dochází k rozvoji svalových atrofií a kontraktur, dysfagie a mnohdy až respirační insuficience. Postižena mohou být i jádra hlavových nervů.
Přibližně 95 % všech případů onemocnění je způsobeno mutací v genu SMN1 (survival motor neuron) a právě pro tyto pacienty je nyní léčba určena. Nejčastějším typem mutace je homozygotní delece exonu 7 nebo exonů 7 a 8 genu SMN1.
Produkt genu SMN1, protein SMN, je nezbytný pro přežití motoneuronů. Avšak přesná patogeneze nemoci zcela jasná není, protože se protein SMN vyskytuje v cytoplazmě a v jádře všech buněk těla, nikoli pouze v motoneuronech, které odumírají. Protein SMN je zapojen do celé řady buněčných funkcí, účastní se sestřihu a transportu mRNA, tvorby růstových faktorů, jeho deficit vede i k dysfunkci mitochondrií a zvýšené náloži oxidačního stresu.
Gen SMN existuje v lidském organizmu ve dvou formách. Nachází se na dlouhém raménku 5. chromozomu – telomerický SMN1 gen a centromerický SMN2 gen. Zdravý jedinec má na každém chromozomu jednu nebo více kopií genu SMN1, tzv. přenašeč má na jednom chromozomu jednu kopii normální a druhou mutovanou a žádné příznaky nemoci nejeví. Nemocný jedinec má mutované kopie obě. Dědičnost nemoci je tedy autosomálně recesivní, a setkají-li se dva tzv. přenašeči, existuje 25% pravděpodobnost, že se jim narodí nemocné dítě (50 % potomků budou přenašeči a 25 % bude zcela zdravých).
Ve většině případů je to tedy tak, že jsou rodiče klinicky zcela asymptomatičtí přenašečia narodí se jim nemocné dítě. Přenašečů je v kavkazské populaci poměrně hodně, udává se prevalence 1 : 38.
Gen SMN2 je genu SMN1 velmi podobný, liší se pouze v pěti nukleotidech, nicméně i takto malá změna vede k tomu, že pouze 10–30 % jeho transkriptů je úplných a funkčních.
Protein produkovaný SMN2 genem tedy nestačí k zabránění nemoci, ale modifikuje její fenotyp. Čím více má pacient kopií genu SMN2, tím jsou příznaky nemoci mírnější.
Stanovení počtu kopií genu SMN2 je již standardní součástí genetického vyšetření nemocných. Poměrně hodně jedinců, přibližně 10–15 % populace, žádnou kopii genu SMN2 nemá a v případě současné mutace genu SMA1 by tento fakt vedl již k prenatálnímu úmrtí.
Klinický obraz
Tradičně se SMA klasifikuje do 3–5 základních typů (typ 0–IV), a to podle věku při začátku potíží a maximálního dosaženého motorického milníku pacienta. Tabulku (tabulka 1) pro úplnost přikládám, avšak toto dělení se při existenci léčby stává stále více pouze orientačním a dosud známé fenotypy se mění. Navíc se při stále přesnější genetické diagnostice bude více uplatňovat i znalost modifikujících genů a samozřejmě počet kopií genu SMN2. Přesnější nyní bude pacienty klasifikovat popisně – podle věku nástupu prvních potíží, typu léčby a době jejího zahájení, počtu kopií genu SMN2 a maximálních dosažených motorických dovedností (pacient ležící, schopný sedu nebo chůze). Nejčastější klinická forma nemoci je SMA typu 1, tvořící přibližně polovinu všech případů. Klinické příznaky jsou patrné buď hned po narození, nebo se rozvíjejí během prvního půl roku života. Dítě je hypotonické, slabě pláče, typická je hyperabdukce v kyčlích. Fenotyp dítěte se označuje jako floppy infant (tedy poddajné dítě). Svalová slabost vede k velmi nápadnému opoždění motorického vývoje, poruše držení hlavičky při slabosti šíjového svalstva. Svalová slabost je více vyjádřena na dolních končetinách, a to proximálně. Svaly postupně atrofují, ale atrofie může být maskována vrstvou podkožního tuku. Motorika rukou může zůstat částečně zachovaná a děti mohou ovládat motorický vozík či počítač. Oslabeny jsou i svaly interkostální, zato bránice je relativně ušetřena, což vede k abdominálnímu typu dýchání. Kolem prvního roku života se objevuje dysfagie vedoucí až k nutnosti zavést perkutánní gastrostomii (PEG).
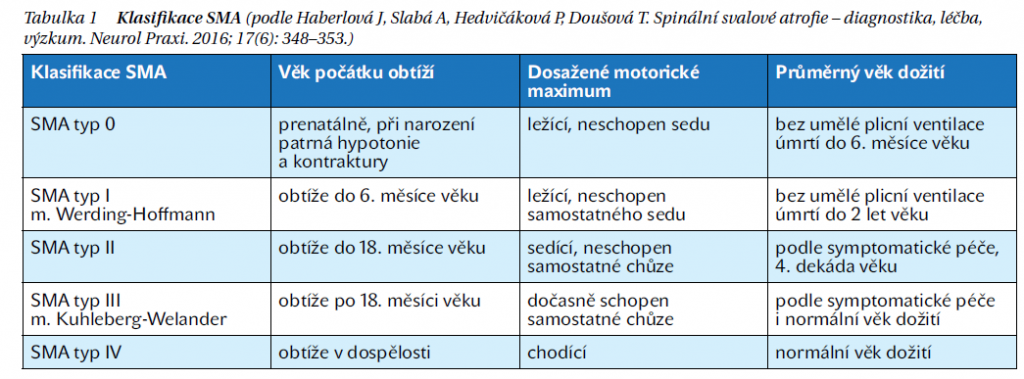
Nápadné a typické je, že tyto děti zachovávají schopnost vytvořit velmi pěkný sociální kontakt. Postupně se rozvíjí noční hypoventilace s hyposaturací, která progreduje i do denní respirační insuficience. Většina dětí (okolo 95 %) bez ventilační podpory umírala do 18 měsíců věku. Respirační insuficience a infekce jsou nejčastější příčinou úmrtí těchto dětí.
Ostatní typy SMA mají fenotyp nemoci mírnější a při existenci novorozeneckého screeningu a kauzální genové léčby bychom takto plně vyvinutý klinický fenotyp neměli v klinické praxi vídat.
Novorozenecký screening
Od 1. ledna 2022 je novorozenecký screening nově rozšířen o vyšetření SMA a také o vyšetření těžké kombinované imunodeficience (SCID). Vyšetření se provádí ze suché kapky krve získané odběrem krve z patičky novorozence. Krev se odebírá 48–72 hodin po narození a odesílá se do screeningové laboratoře, kde je testována na dnes už tedy 20 vrozených vzácných léčitelných onemocnění. Vyšetření na SMA a SCID je dobrovolné a je nutný písemný souhlasu rodičů. Lze ho provést v každé porodnici a výsledek je k dispozici přibližně za 14 dní. Pro účastníky veřejného zdravotního pojištění v ČR je vyšetření plně hrazeno.
Diagnostika
Většina nemocných by tedy nyní měla být diagnostikována právě pomocí novorozeneckého screeningu. K diagnostice zbývají nemocní, kteří se narodili před zavedením screeningu, dále děti, jejichž rodiče screening odmítli nebo odmítnou, a pacienti s jiným typem SMA, než je SMA s mutací genu SMN1 (těch je přibližně 5 % a níže uvedená léčba pro ně není určena).
Základem správné diagnózy je pečlivý odběr anamnézy a objektivní neurologické vyšetření. Potom jsou provedeny laboratorní odběry, kde může být zvýšená hladina kreatinkinázy (avšak maximálně do pětinásobku normy). Následuje genetické vyšetření.
Zvláště u starších pacientů se může ještě před genetickým vyšetřením provést elektromyografie (EMG).
Pokud genetické vyšetření SMA nepotvrdí, diagnostický proces pokračuje většinou doplněním MR mozku a míchy, MR svalů s ev. provedením svalové biopsie. Zvláště právě formy SMA s mutací jinou než v genu SMN1 mohou být klinicky velmi heterogenní a diagnosticky náročné. Nedávno jsme vyšetřovali dospělou ženu a její dceru pro poruchu chůze, která imponovala jako myopatická. Nakonec se prokázalo, že se jedná o vzácný typ SMA s mutací genu pro protein dynein.
Dostupná léčba
Nusinersen (Spinraza)
Prvním kauzálním lékem zavedeným do praxe byl lék nusinersen (Spinraza). Nusinersen zvyšuje tvorbu chybějícího SMN proteinu tím, že moduluje transkripci RNA genu SMN2.
V Evropě byl schválen v květnu roku 2017 a týž rok léčbu obdržel první český pacient.
V České republice je nyní hrazen pro všechny typy nemoci s mutací genu SMN1 a především i pro všechny věkové skupiny pacientů se SMA.
Podává se intratekálně, tedy pomocí lumbální punkce, jelikož nepřechází přes hematoencefalickou bariéru. Musí být podáván pravidelně. Po první dávce (1 injekční lahvička, tj. 12 mg) následují další tři dávky po 2, 4 a 9 týdnech a následně jedna dávka každé 4 měsíce, a to celoživotně. Jeho nevýhodou je právě nutnost opakovaných lumbálních punkcí.
Nejčastějšími nežádoucími účinky jsou postpunkční potíže (především bolest hlavy a nauzea). Ojediněle, řádově v jednotkách případů, byl popsán rozvoj komunikujícího hydrocefalu, avšak jasná souvislost s léčbou prokázána není.
Ze všech tří dostupných léčiv je s nusinersenem nejvíce klinických zkušeností, odléčeno je více než 10 000 pacientů.
Risdiplam (Evrysdi)
Risdiplan funguje na obdobném principu jako nusinersen, tedy moduluje transkripci RNA genu SMN2. Na rozdíl od nusinersenu se ale užíá perorálně. V České republice je indikován pro všechny pacienty se SMA (s vazbou na gen SMN1), kteří jsou starší dvou měsíců a mají počet kopií genu SMN2 ≤ 4. Doporučená denní dávka se stanovuje podle věku a tělesné hmotnosti pacienta, avšak maximálně je to 5 mg.
Ve studiích na zvířatech byla zjištěna embryofetální toxicita a u pacientů ve fertilním věku je během léčby nezbytné užívat vysoce účinnou antikoncepci, a to i nejméně 1 měsíc po poslední dávce v případě žen a 4 měsíce po poslední dávce v případě mužů. Mezi nejčastější nežádoucí účinky patří horečka, kožní vyrážka, aftózní vřídky v ústech, bolest hlavy, artralgie a močové infekce.
Onasemnogen abeparvovek (Zolgensma)
Třetí kauzální lék, v Evropě registrovaný od května 2020, slouží k jednorázové léčbě. Podává se formou cca hodinové infuze. Jeho indikační kritéria jsou ale přísnější. V současnosti je v ČR schválen pouze pro část dětských SMA pacientů, a to pro děti od narození do 3 let (maximální stáří léčeného pacienta je den před třetími narozeninami) a do hmotnosti 13,5 kg.
Princip léčby je jiný než u předchozích dvou preparátů. Lék nahrazuje chybějící gen SMN1 genem syntetickým, který je do organizmu přenesen virovým vektorem – adenoasociovaným virem typu 9 (AAV9). Způsob podání spojený s virovým vektorem s sebou nese jistá rizika a specifika léčby. Podání léčiva je provázeno imunitní odpovědí organizmu na kapsid AAV9, což může být provázeno elevací jaterního souboru, zvýšením troponinu I či trombocytopenií.
Před léčbou je pacient premedikován imunomodulační léčbou kortikoidy a v kortikoterapii se pokračuje i 2 měsíce po podání léku. Nezbytná je pravidelná kontrola jaterních testů, a to nejméně po dobu 3 měsíců. Sledována musí být i hladina trombocytů a troponinu I.
Z nežádoucích účinků se kromě zmíněné hepatotoxicity, trombocytopenie a elevace troponinu vyskytuje také horečka a zvracení.
Uvedená léčiva zásadním způsobem mění prognózu pacientů se SMA
Nejdůležitější prediktor nejlepšího efektu je včasnost zahájení léčby. Existuje přímá korelace mezi časným začátkem léčby, počtem zachovaných motorických neuronů a efektem léčby. Ihned po stanovení diagnózy jsou léčení pacienti se SMA typu 1 většinou schopni sedu nebo i stoje, pacienti se SMA typu 2 mohou chodit s dopomocí, nemocní se SMA typu 3 si schopnost chůze zachovají místo toho, aby ji ztratili. I přes zlepšení pohybových schopností ale může dojít ke zhoršení respiračních funkcí a polykání. Zásadní pozitivní efekt by zde měl přinést novorozenecký screening a léčba již v presymptomatickém stadiu. Takto léčené dítě by se teoreticky mělo vyvíjet jako zcela zdravé, avšak zda efekt léčby vydrží opravdu celý život, dnes zatím nevíme. Nyní jsou k dispozici data o účinku léčby trvajícím 7 let a tento efekt zatím přetrvává.
U pacientů s pokročilejší formou nemoci je hlavním efektem zastavení progrese, ale k mírnému zlepšení motoriky může také dojít. Je tedy důležité, aby pacienti a jejich rodiny neměli od léčby nereálná očekávání. Pro nemocné má však i zdánlivě malé zlepšení motorických dovedností významný vliv na zlepšení kvality života.
Na jaře roku 2022 jsme v rámci brněnského neuromuskulárního kongresu viděli dojemné příběhy brněnských a pražských pacientů, kterým léčba život velmi výrazně změnila k lepšímu. Byl prezentován příběh muže, jemuž zlepšení pohybových dovedností umožnilo udělat si řidičský průkaz, či příběh chlapce, kterému přestala přepadávat hlava, a oba se tak stali mnohem více soběstačnými.
Závěr
V případě SMA jsme svědky velikého přelomu v léčbě dosud neléčitelné a často fatální nemoci. Takový převrat a objev v léčbě přináší naději do budoucna i pro naše ostatní neurologické pacienty s dosud neléčitelnými neurodegenerativními onemocněními.
Věřím, že nás v neurologii čekají optimistické časy, kdy se z neléčitelných nemocí začnou pomalu stávat onemocnění léčitelná a my budeme schopni pomoci i těm pacientům, u kterých to dnes příliš nedokážeme. Příkladem může být lék Tofersen, cílený na specifickou podskupinu nemocných s amyotrofickou laterální sklerózou (ALS) s mutací genu pro superoxid dismutázu 1 (SOD 1), který se rovněž jeví jako velmi nadějný.
MUDr. Pavlína Hemerková
prof. MUDr. Martin Vališ, Ph.D., FEAN
Neurologická klinika LF UK a FN Hradec Králové