Neuromyelitis optica (NMO, známá jako Devicova nemoc) a poruchy jejího širšího spektra (NMOSD – neuromyelitis optica spectrum disorder) je vzácné a závažné autoimunitní onemocnění centrálního nervového systému charakterizované záněty očního nervu, míchy a méně často mozkového kmene. Objevem protilátek proti akvaporinu 4 (AQP4-IgG/NMO-IgG) se rozšířilo spektrum klinických příznaků tohoto onemocnění a pomohlo definitivně odlišit NMO od roztroušené sklerózy.
Mezi hlavní léčebné strategie patří agresivní léčba relapsů (vysokodávkované steroidy či plazmaferéza) následovaná zahájením chronické terapie, mezi léky první volby dnes řadíme monoklonální protilátky ekulizumab, inebilizumab, satralizumab nebo rituximab.
Stanovení AQP4-IgG v séru spolu s magnetickou rezonancí mozku a míchy mají nezastupitelný význam v diagnostice tohoto onemocnění již v okamžiku první manifestace choroby. Typickým nálezem jsou míšní ložiska přesahující délku tří míšních segmentů. Důležité informace přináší i optická koherentní tomografie (OCT). Onemocnění vede k těžkému neurologickému deficitu, proto je zásadní časné stanovení diagnózy a nasazení adekvátní terapie, která může změnit nepříznivou prognózu pacienta.
NMO byla poprvé popsána Eugenem Devicem v 19. století a dříve byla považována za agresivní podtyp roztroušené sklerózy. V roce 2004 V. Lennová et al. objevili protilátky proti akvaporinu 4 (AQP4-IgG/NMO-IgG), specifické pro tuto chorobu. Cílovým antigenem těchto protilátek je akvaporin 4 (AQP4), který je exprimován zejména astrocyty. Patří do rodiny transmembránových vodních kanálů, které se významně podílejí na vodní homeostáze a jsou součástí hematoencefalické bariéry. AQP4 zprostředkovává transport vody přes plazmatickou membránu, selektivní destrukce a dysfunkce astrocytů vede dále k demyelinizaci a ztrátě neuronů. NMO se definitivně vyčlenila jako samostatná nozologická jednotka a dnes ji řadíme mezi protilátkově zprostředkovaná autoimunitní onemocnění.
Epidemiologie NMO
Před objevem protilátek proti AQP4 byla NMO považována za onemocnění s extrémně vzácným výskytem u indoevropské populace v našich geografických šířkách, onemocnění bylo v Evropě zřejmě poddiagnostikováno a pacienti byli často vedeni pod diagnózou roztroušené sklerózy.
Údaje o incidenci a prevalenci jsou limitované a vycházejí zvláště z národních registrů. Prevalence v Evropě se odhaduje na 0,5– 4,4 na 100 000 obyvatel. Odhaduje se, že toto onemocnění tvoří asi 1,5 % získaných demyelinizačních onemocnění CNS u evropské populace. Naopak u asijské populace je výskyt mnohem vyšší a tvoří asi 20– 40 % onemocnění v této skupině. Častěji onemocní ženy (poměr se pohybuje od 2,8– 9 : 1), medián věku začátku onemocnění je okolo 40 let. První manifestace nemoci může být ale prakticky v kterémkoli věku, včetně první a sedmé dekády života.
Patogeneze
Hlavním patofyziologickým mechanizmem je navázání AQP4-IgG na cílový antigen, kterým je astrocyty exprimovaný akvaporin 4, což vede zejména k aktivaci komplementu a přímému poškození astrocytů vznikem terminálního lytického komplexu. Štěpné produkty aktivované komplementové kaskády fungují chemotakticky pro leukocyty, hl. neutrofily a eozinofily, které se spolupodílejí na destrukci nervové tkáně uvolněnými proteázami a volnými radikály. Zvýšené sérové koncentrace C3a složky komplementu korelují s neurologickým deficitem a aktivitou choroby. Akutní NMO léze je charakterizována splývající a/ nebo fokální perivaskulární demyelinizací, prominentní infiltrací makrofágů, ztrátou axonů, nekrózami šedé i bílé hmoty míchy, provázené ztrátou astrocytů.
Změny nacházíme v šedé i bílé hmotě. Jedná se tedy o protilátkami zprostředkované onemocnění a z pohledu patologa je NMO primární astrocytopatií se sekundárně indukovanou demyelinizací.Základním rozdílem v patogenezi NMO v porovnání s roztroušenou sklerózou (RS) je primární postižení astrocytů po navázání protilátek v oblasti hematoencefalické bariéry (HEB), systémová aktivace komplementu a infiltrace granulocytů způsobující primární postižení. Demyelinizace a ztráty neuronů jsou pak sekundárním projevem indukovaného zánětu.
Klinický obraz
Klinický obraz vychází z postižení zrakového nervu, míchy a mozkového kmene. V 90 % případů se setkáváme s relaps remitentním průběhem (střídání atak s remisemi). Délka remise je variabilní od několika týdnů po několik let. U méně než 10 % pacientů je průběh monofázický, podle Devicova konceptu souběžně probíhající myelitidy a optické neuritidy. Díky objevu protilátek proti AQP4 se rozšířily klinické příznaky tohoto onemocnění. Zejména se jedná o příznaky při postižení mozkového kmene, hypothalamu nebo sluchu.
Optická neuritida (ON) má u pacientů s NMO a NMOSD závažnější prognózu než u RS. Projevuje se bolestí za okem, výpadky zorného pole, změnou barvocitu nebo snížením zrakové ostrosti. Tíže postižení zrakového nervu, popřípadě jeho bilaterální postižení, by nás mělo vést k úvaze o zánětlivém postižení v rámci onemocnění NMO. Dále je typický poměrně rychlý rozvoj s výrazným otokem na papile, těžký průběh bez úplné úpravy, eventuálně amauróza, kterou vyvine asi 22 % pacientů po první atace. U některých pacientů může dojít k rozvoji amaurózy během několika hodin. Zde pak může být mylně stanovena diagnóza přední ischemické neuropatie optiku i vzhledem k věku, protože počátek onemocnění v páté dekádě života a později je poměrně častý.
Kmenové příznaky jsou často opomíjené a jsou vyjádřeny asi u čtvrtiny pacientů, mnohdy v úvodu onemocnění. Typicky se jedná o nauzeu a vomitus doprovázené singultem, často v souvislosti s lézí v oblasti area postrema. Vzácnější jsou ztráta sluchu, vestibulární syndrom, diplopie, neuralgie trigeminu a parézy hlavových nervů. V důsledku postižení dechového centra v oblasti medulla oblongata může dojít k neurogenně podmíněnému respiračnímu selhání, se kterým se setkáváme zejména u pacientů se souběžnou rozsáhlou myelitidou oblasti krční a hrudní míchy. Postižení hypothalamu se může projevit polyurií, hypersomnií, hypotermií, hypotenzí a obezitou. Příznaky bývají doprovázeny T2 hyperintenzitami na magnetické rezonanci (MR) mozku v oblasti hypothalamu.
Myelitida má také závažnější průběh než u RS. Probíhá často pod obrazem para- či kvadruparézy nebo plegie s hranicí čití a sfinkterovými poruchami. Rozvoj příznaků může být rychlý, imitující až cévní etiologii. Inkompletní míšní léze diagnózu NMO nevylučuje, naopak se s ní setkáváme u pacientů, kteří užívají imunosupresivní léčbu pro prodělané ON nebo mají jiné autoimunitní onemocnění (např. systémový lupus erythematodes). Radikulární bolest, paroxysmální tonické velmi bolestivé svalové spasmy a decharge electrique (Lhermittův příznak) se vyskytují u jedné třetiny relaps remitentních pacientů, ale nenacházíme je u pacientů s monofázickým průběhem NMO. Na magnetické rezonanci míchy typicky vídáme tzv. longitudinálně extenzivní transverzální myelitidu (LETM), jejíž délka dosahuje nebo přesahuje tři obratlové segmenty v T2W obrazech. Klinická úprava těžkého neurologického deficitu závisí na zahájení adekvátní terapie, ale často zůstává výrazné reziduum. V kohortě pacientů s mediánem sledování 75 měsíců s počátkem onemocnění nad 46. rokem života bylo více než 40 % odkázáno na invalidní vozík. S vyšším věkem počátku onemocnění roste riziko trvalého motorického deficitu.
U pacientů s NMO se často setkáváme s výskytem dalších autoimunitních onemocnění, jako je autoimunitní thyroiditida, systémový lupus erythematodes (SLE), Sjögrenův syndrom (SS), diabetes mellitus 1. typu a myasthenia gravis. Někdy se u pacientů setkáváme s určitou klinickou symptomatologií, která odpovídá příznakům typickým např. pro SLE (artritida, leukopenie), ale nenaplní kritéria pro dané onemocnění.
Těhotenství a období po porodu je u pacientek s NMO/ NMOSD velmi rizikové. Jakkoli je celosvětově v literatuře popsáno několik desítek případů, výsledná data hovoří o rizikovosti zejména v prvním a třerím trimestru a období po porodu.
Pomocné vyšetřovací metody
Stanovení diagnózy NMO a onemocnění jejího širšího spektra (NMO SD)
V rámci stanovení diagnózy vycházíme z klinického obrazu a výsledků pomocných vyšetřovacích metod. V roce 2015 byla na základě konsenzu expertů publikována revidovaná Wingerchukova diagnostická kritéria (tabulka 1), která pomohla k rychlejší diagnostice a léčbě pacientů po první atace či hraničních případů. Tato kritéria upřesňují termín neuromyelitis optica spectrum disorders (NMOSD) a upřesňují dvě hlavní skupiny pacientů: (1) s pozitivitou protilátek proti AQP4, u kterých stačí k průkazu diagnózy přítomnost jednoho ze šesti hlavních klinických příznaků; (2) s negativitou protilátek, kde se vyžaduje přítomnost alespoň dvou a více hlavních klinických příznaků, přičemž jeden musí být typický pro NMOSD (ON, LETM nebo area postrema).
U AQP4-IgG séronegativních pacientů s NMO je diferenciální diagnóza širší, je nutné vyloučit jiná autoimunitní onemocnění, infekce, tumory, metabolické příčiny. Část séronegativních pacientů s NMO může mít pozitivní protilátky proti myelinovému oligodendrocytárnímu proteinu (MOG-IgG) nebo alfa podjednotce glycinového receptoru.
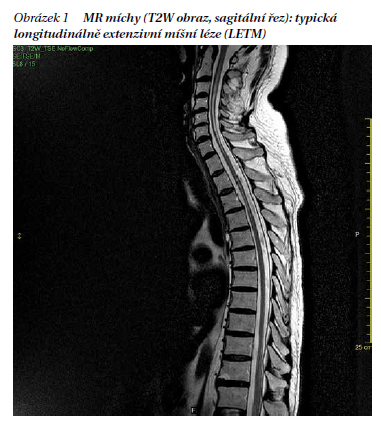
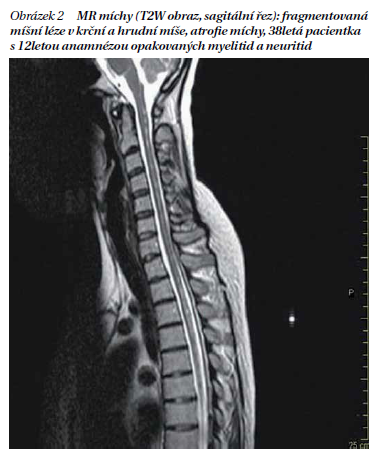
Magnetická rezonance mozku a míchy je klíčovým zobrazovacím vyšetřením v diagnostice NMO, doporučeno je i podání kontrastní látky. U pacientů s myelitidou nacházíme na magnetické rezonanci typicky ložisko dosahující nebo přesahující tři obratlové segmenty a hovoříme o tzv. longitudinálně extenzivní myelitidě (LETM), která mívá v akutním stadiu na T2 vážených obrazech typicky vřetenovitý tvar (obrázek 1), postihuje šedou i bílou hmotu a může se po podání kontrastní látky sytit. Ložiska se typicky vyskytují v krční a hrudní míše. S odstupem od akutního stadia a po léčbě kortikoidy dochází k fragmentaci léze (obrázek 2) a u některých pacientů může dojít k jejímu vymizení nebo je přítomno jen syringomyelické rozšíření centrálního kanálku. Časem se vyvíjí výrazná atrofie, zejména v oblasti hrudní míchy. LETM není pro NMO specifická, v praxi vídáme u pacientů na MR míchy také myelitidy kratší než 3 obratové segmenty. Krátká léze míchy tedy NMO nevylučuje.
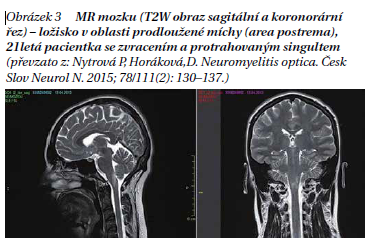
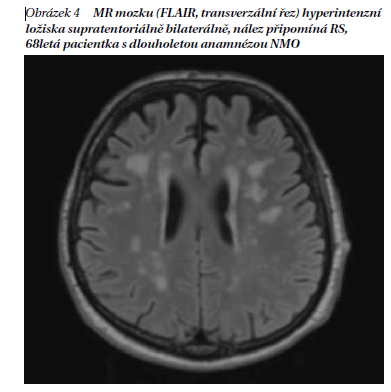
Původní předpoklad, že pacienti s NMO mají na MR mozku negativní nález, je dnes již vyvrácen. Z různých studií vyplývá, že asi 50 % pacientů s NMO má na MR nález již na počátku onemocnění a pravděpodobnost změn na MR mozku koreluje s délkou trvání onemocnění. Nález je nespecifický, může napodobovat RS (obrázek 3), nicméně atypická lokalizace, často ve spojitosti s atypickými projevy, by měla upozornit na možnost NMO. Změny na MR mozku u NMO pacientů odpovídají expresi AQP4. Typické oblasti, které bývají postiženy a které by nás měly spolu s klinickým obrazem vést ke zvážení diagnózy NMO, jsou následující:
– v periependymální oblasti Sylviova akveduktu, IV. a III. komory;
– v oblasti prodloužené míchy často navazující na ložisko míšní, typicky s postižením area postrema (obrázek 4);
– v oblasti postranních komor a corpus callosum (většinou rozsáhlejší ložiska než u RS, někdy popisován neostře ohraničený skvrnitý enhancement);
– v oblasti pyramidové dráhy (zadní raménko capsula interna), horní pedunkuly mozečku,
– rozsáhlé léze v oblasti bílé hmoty supratentoriálně (vzácněji).
Stanovení protilátek proti AQP4 (AQP4-IgG) v séru pacienta je základní vyšetření v rámci stanovení diagnózy. Zlatým standardem je stanovení protilátek pomocí metody nepřímé imunofluorescence nebo průtokové cytometrie. Specificita protilátek je prakticky 100 %, senzitivita 80 %, z toho vyplývá, že negativita protilátek nevylučuje diagnózu. Pokud trvá klinické podezření, pak je vhodné při negativním výsledku odběr opakovat s odstupem několika týdnů, nejlépe jinou laboratorní metodou. U většiny pacientů přetrvává sérová pozitivita při opakovaných odběrech, ke snížení koncentrace může dojít při léčbě nebo v remisi.
Vyšetření mozkomíšního moku nám pomáhá zejména v diferenciální diagnóze. V atace nacházíme porušenou hematoencefalickou bariéru (zvýšená koncentrace bílkoviny) s oligo- či pleiocytózou, kde mohou být přítomny kromě lymfocytů i eozinofilní a neutrofilní granulocyty. Vždy doplňujeme sérologii a PCR k vyloučení infekční etiologie obtíží pacienta. Asi u 30 % pacientů mohou být pozitivní oligoklonání pásy (oligoclonal bands – OCB), popř. se můžeme setkat s obrazem shodného výskytu pásů v séru a likvoru. Právě negativita OCB v likvoru s podezřením na RS by nás měla vést k úvaze o NMO, ale přítomnost OCB diagnózu nevylučuje.
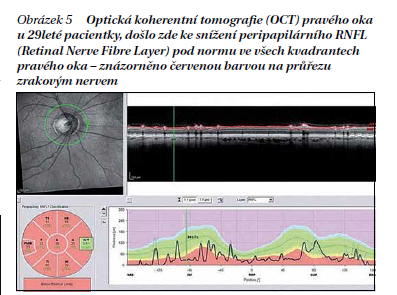
U pacientů s optickou neuritidou vyšetřujeme vizuální evokované potenciály (VEP) nebo optickou koherentní tomografii (OCT). Vizuální (zrakové) evokované potenciály vykazují abnormality charakteru prodloužení vlny P100, redukci amplitudy nebo chybění potenciálu, léze je většinou prechiasmatická. Optická koherentní tomografie (OCT) je neinvazivní zobrazovací metoda, která zobrazuje příčné řezy sítnicí. Pomocí OCT měříme tloušťku retinálních nervových vláken, tzv. RFNL (retinal nerve fiber layer thickness). U NMO je typické výrazné postižení ve všech nebo většině kvadrantů (obrázek 5), u RS je častěji pokles RNFL pouze v temporálních kvadrantech.
Stanovení diagnózy NMO a onemocnění jejího širšího spektra (NMO SD)
V rámci stanovení diagnózy vycházíme z klinického obrazu a výsledků pomocných vyšetřovacích metod. V roce 2015 byla na základě konsenzu expertů publikována revidovaná Wingerchukova diagnostická kritéria (tabulka 1), která pomohla k rychlejší diagnostice a léčbě pacientů po první atace či hraničních případů. Tato kritéria upřesňují termín neuromyelitis optica spectrum disorders (NMOSD) a upřesňují dvě hlavní skupiny pacientů: (1) s pozitivitou protilátek proti AQP4, u kterých stačí k průkazu diagnózy přítomnost jednoho ze šesti hlavních klinických příznaků; (2) s negativitou protilátek, kde se vyžaduje přítomnost alespoň dvou a více hlavních klinických příznaků, přičemž jeden musí být typický pro NMOSD (ON, LETM nebo area postrema).

U AQP4-IgG séronegativních pacientů s NMO je diferenciální diagnóza širší, je nutné vyloučit jiná autoimunitní onemocnění, infekce, tumory, metabolické příčiny. Část séronegativních pacientů s NMO může mít pozitivní protilátky proti myelinovému oligodendrocytárnímu proteinu (MOG-IgG) nebo alfa podjednotce glycinového receptoru.
Léčebé přístupy
Zatím nemáme léky, které by nemoc zcela vyléčily. Za poslední roky ale došlo k významnému posunu v poznání patogenetických mechanizmů, a tím i možností cílené léčby, která nemoc u velké části pacientů dobře stabilizuje. Vzhledem k tomu, že u přirozeného průběhu nemoci hrozí již časně těžká invalidita, která je většinou následkem proběhlého relapsu, tak je maximum úsilí směrováno právě na agresivní léčbu akutního zhoršení a následně na dlouhodobou/ trvalou prevenci dalších atak. Nedílnou součástí péče je i léčba symp¬tomatická (sfinkterové dysfunkce, neuropatická bolest, spasticita atd.) a prevence infekcí. Léky používané v chronické léčbě RS u NMOSD selhávají, a některé mohou dokonce chorobu zhoršit (fingolimod, natalizumab, interferony), což je dáno rozdílnou patogenezí obou chorob. Vzhledem k tomu, že se jedná o relativně vzácné onemocnění s vysokým rizikem časné invalidity, je provádění randomizovaných, placebem kontrolovaných studií velmi obtížné. Léčbu je možné rozdělit na terapii relapsu, léčbu chronickou a symptomatickou.
Léčba relapsu onemocnění je zahajována vysokodávkovanými kortikoidy – methylprednisolon v dávce 5 g (1 g/ den) s navazujícím pozvolným snižováním perorálních kortikoidů. Pokud efekt není dostatečný, je doporučeno bez prodlení zahájit sérii plazmaferéz (obvykle v počtu 5– 7 výkonů obden). U pacientů s těžkým relapsem a známou diagnózou, kdy byl již v minulosti dobrý efekt plazmaferézy, je doporučeno zahájit jako první přímo sérii plazmaferéz, možno použít i střídání plazmaferéz a 1 g methylprednisolonu obden. U plazmaferéz doporučujeme provádění výkonů pouze na pracovištích se zkušenostmi s těmito výkony. V případě intolerance předchozích režimů je další variantou podání intravenózních imunoglobulinů (IVIG) nebo imunoadsorpce.
Úspěšná chronická léčba vede k redukci frekvence relapsů a jejich tíže. V případě definitivní diagnózy NMO je doporučeno ihned zahájit chronickou terapii s cílem prevence dalších devastujících atak. V minulosti byli pacienti léčeni imunosupresivní terapií, tato terapie byla v posledních letech nahrazena léčbou více cílenou na patogenezi NMO. Tyto léky jsou zaměřeny přímo na autoimunitní kaskádu specifickou pro NMO. Jedná se o léky, které cílí na interleukin 6 (satralizumab, tocilizumab), inhibují komplement (ekulizumab) nebo vedou k depleci B lymfocytů (rituximab, inebilizumab). Od roku 2019 byla FDA (Úřad pro kontrolu potravin a léků) schválena léčba třemi léky, jedná se o ekulizumab, inebilizumab a satralizumab, na všechny proběhly radomizované studie. Všechny tři léky jsou velmi nákladné, proto řada lékařů používá v léčbě i off-label rituximab a tocilizumab. Většina léků se podává ve formě intravenózních infuzí.
Ekulizumab je humánní protilátka, která blokuje aktivaci komplementové kaskády vazbou na C5 složku komplementu. Léčba je spojená s vyšším rizikem bakteriálních infekcí hlavně meningokokové nákazy, proto se doporučuje před léčbou očkování proti meningokokům a v průběhu léčby antibiotická profylaxe. Ekulizumab se dále používá v léčbě refrakterní myasthenia gravis, paroxysmální noční hemoglobinurie a hemolyticko-uremického syndromu. Častými nežádoucími účinky jsou bolest hlavy, infekce a anémie.
Inebilizumab je humánní protilátka, která se váže na CD 19 povrchový antigen B buněk a vede k depleci B lymfocytů. Před léčbou je vhodný screening na hepatitidy a tuberkulózu, častými nežádoucími účinky jsou bolesti hlavy, uroinfekce a infuzní reakce.
Satralizumab a tocilizumab jsou humánní monoklonální protilátky, které se vážou na interleukin 6. Interleukin 6 podporuje aktivaci autoreaktivních T buněk, prodlužuje přežití B buněk produkujících AQP4-Ab a narušuje integritu hematoencefalické bariéry. Tocilizumab se podává intravenózně a používá se v léčbě revmatoidní artritidy. Satralizumab je ve formě subkutánních injekcí.
Rituximab je chimérická myší/ lidská anti‑CD20 monoklonální protilátka. Při jeho podání dochází k depleci B-lymfocytů. V praxi používáme terapeutické schéma 1 000 mg i. v. v odstupu 14 dní. Ve skupině 30 pacientů léčených rituximabem po dobu 5 let dosáhlo 93 % pacientů stabilizace EDSS skóre nebo došlo k mírnému zlepšení neurologického nálezu. Podání další terapeutické dávky (stejné terapeutické schéma) je doporučeno v okamžiku před znovuobjevením se CD19 pozitivních buněk (vyšetření průtokovou cytometrií). Tento interval je u pacientů individuální, ale nejčastěji se pohybuje mezi 6–8 měsíci. Z našich zkušeností je u některých pacientů monoterapie rituximabem nedostačující, proto kombinujeme s nízkou dávkou kortikoidů.
Lékem první volby bylo v minulosti imunopsuresivum azathioprin v monoterapii či kombinaci kortikoidy. Azathioprin má doporučenou denní dávku 2,5– 3 mg/ kg tělesné hmotnosti/ den. V úvodu je doporučeno kombinovat s vyšší dávkou orálních steroidů (např. prednisonem v dávce až 1 mg/ kg/ den s velmi pomalým snižováním dávky), dlouhodobě je pak možné kombinovat s nižší dávkou prednisonu (5– 15 mg/ den). Před nasazením azathioprinu je třeba vyšetřit aktivitu enzymu TPMT (thiopurinmetyltransferáza), v případě nedostatečné aktivity je doporučeno azathioprin nepodávat pro vysoké riziko dřeňového útlumu. Dalšími imunosupresivními léky, které se používaly v léčbě NMO jako léky 2. volby, jsou mitoxantron, mykofenolát mofetil, methrotrexát, cyklosporin A a cyklofosfamid.
Všechny léčebné režimy vyžadují dodržování bezpečnostních doporučení, týkajících se např. kontroly krevního obrazu, jaterních funkcí a dalších parametrů. V současné době ale vzhledem k účinnosti léčby upřednostňujeme cílenou léčbu uvedenou výše nad imunosupresivy.
Délka terapie u pacientů zatím není jasná, mělo by být dosaženo stabilizace choroby a léčba by podle dostupných studií měla být ponechána aspoň 5 let.
Závěr
NMO a poruchy jejího širšího spektra se vyskytují v naší klinické praxi poměrně vzácně, ale nesmíme na tuto možnost v diferenciální diagnostice zapomínat. Důležité je hlavně odlišení NMO a RS vzhledem k odlišné terapii. Diagnostika se výrazně zjednodušila díky vyšetření AQP4- IgG, které je dnes dobře dostupné i v České republice. Vzhledem k riziku časné těžké invalidity je důležité určit diagnózu v okamžiku první manifestace onemocnění, protože pouze agresivní léčba relapsu a následná dlouhodobá léčba mohou změnit negativní prognózu pacienta.
MUDr. Jana Pavlíčková, Ph.D., MUDr. Petra Nytrová, Ph.D.
Neurologická klinika a Centrum klinických neurověd, 1. LF UK a VFN Praha